AUCell Tutorial
In this notebook, we introduce a tutorial to calculate the AUCell score with our library: bioalpha.
The implementation of bioalpha.sc.tl.aucell is based on pySCENIC.
1. Prepare
This notebook uses gene set of mouse downloaded from GSEA and a 10k cell single-cell ma-seq dataset obtained from 10xGENOMICS.
Dataset: 10k Heart Cells from an E18 mouse (v3 chemistry)
Gene sets: m1.all.v2023.1.Mm
1.1. Load required library
import bioalpha
1.2. Download data
!wget -P ./data/enrichment/ https://data.broadinstitute.org/gsea-msigdb/msigdb/release/2023.1.Mm/m1.all.v2023.1.Mm.json
!wget -P ./data/datasets/ https://cf.10xgenomics.com/samples/cell-exp/3.0.0/heart_10k_v3/heart_10k_v3_raw_feature_bc_matrix.h5
1.3. Read data
adata = bioalpha.sc.read_10x_h5("./data/datasets/heart_10k_v3_raw_feature_bc_matrix.h5")
adata.var_names_make_unique()
adata
AnnData object with n_obs × n_vars = 6794880 × 31053
var: 'gene_ids', 'feature_types', 'genome'
BioAlpha supports read .json file enrichment downloaded from GSEA.
ENRICHMENT_PATH = "./data/enrichment/m1.all.v2023.1.Mm.json"
# gene_sigs = bioalpha.sc.read_gene_signatures(ENRICHMENT_PATH)
Or you can save gene signatures directly to adata.uns["gene_sigs"] by passing argument to parameter adata.
bioalpha.sc.read_gene_signatures(ENRICHMENT_PATH, adata=adata)
2. Basic filter
bioalpha.sc.pp.filter_cells(adata, min_genes=200)
bioalpha.sc.pp.filter_genes(adata, min_cells=3)
3. Calc AUCell
In this notebook, we set normalize = True in order to visualize heatmap easier (which will introduce in next step).
bioalpha.sc.tl.aucell(adata, normalize=True, gene_sigs="gene_sigs")
If your have your own gene signature, pass argument to parameter gene_sigs.
# bioalpha.sc.tl.aucell(adata, gene_sigs=gene_sigs, normalize=True)
4. Plot AUCell
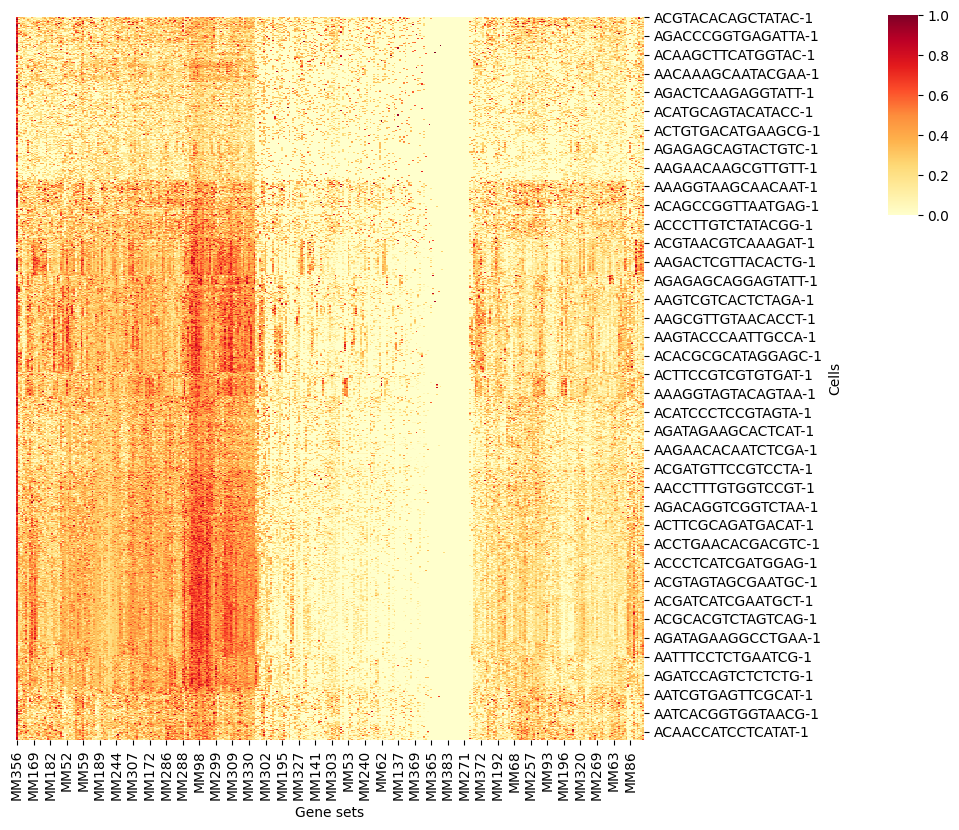
4.1. Heatmap
Note that we only show the first 1000 cells in each group.
bioalpha.sc.pl.aucell_heatmap(adata, cmap="YlOrRd", dendorgram=False)
WARNING: groups size is larger than 1000, only use first 1000 cells
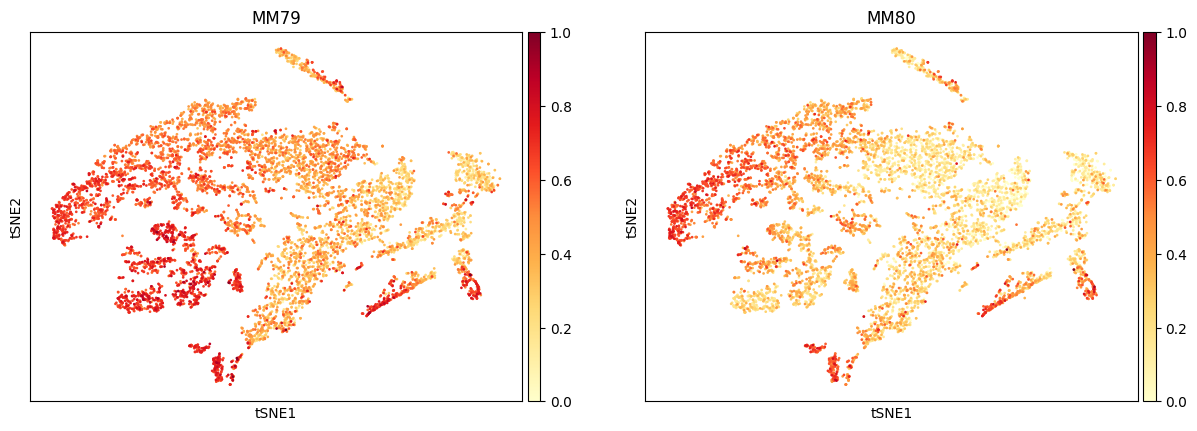
4.2. Plot with define gene sets
We now want to draw the AUCell score on a t-SNE or UMAP coordinate.
# Get 2D-array of t-SNE
bioalpha.sc.pp.pca(adata, n_comps=50)
bioalpha.sc.tl.tsne(adata)
bioalpha.sc.pl.aucell(adata, gene_sets=['MM79', 'MM80'], basis="tsne")